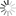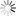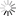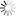新闻网讯 1月28日,《物理评论快报》(Physical Review Letters)在线刊发了能源学院冯光教授课题组关于恒电势分子模拟方法的最新研究成果。论文题目为“考虑电子溢出效应的双电层恒电势模拟(Constant-potential modeling of electrical double layers accounting for electron spillover)”,华中科技大学为唯一通讯单位,博士生王祯祥与博士后陈明为共同一作,通讯作者为冯光教授。
电极与电解液形成的固液界面双电层在电化学体系中广泛存在。从分子层面探究双电层的微观结构与动态响应,来揭示电化学界面的形成机制,是研究电化学体系的关键所在。恒电势分子模拟是研究双电层的重要工具;然而,在金属电极-水溶液界面基准体系上,常规恒电势方法预测的微分电容,通常比实验小一个量级,且随电压变化不大——与实验给出的钟形电容曲线定性不同,这成为了利用恒电势分子模拟方法研究双电层体系的热点与难点。
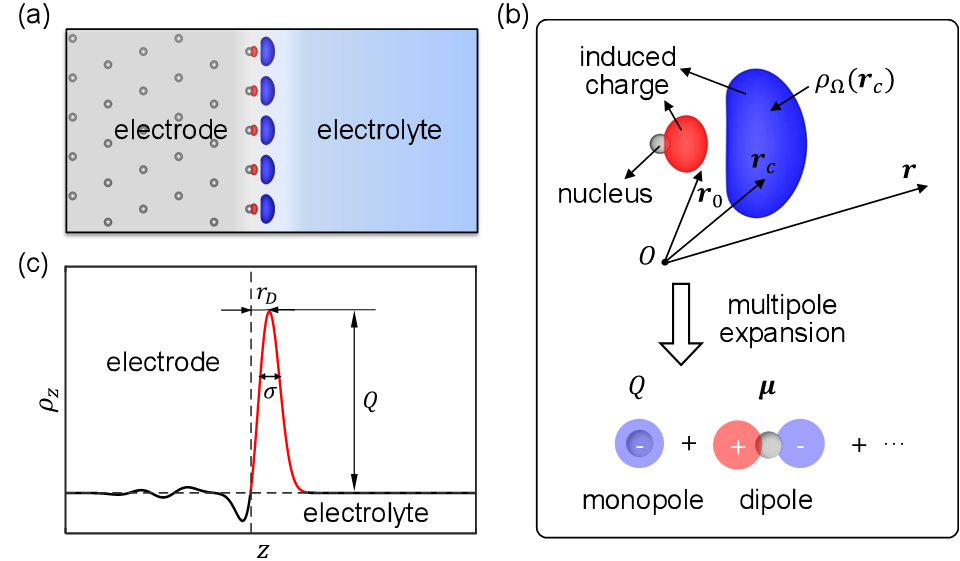
图为考虑电子溢出效应分子模拟方法的示意图(a)双电层界面的电极诱导电荷;(b)诱导电荷的多极矩展开;(c)诱导电荷分布的示意图。红色、蓝色等值面分别代表正电和负电。
针对上述问题,研究团队开发了考虑电极电子溢出效应的恒电势分子动力学模拟方法,深入研究了金电极-水溶液界面的基准体系,解析了其微分电容和界面结构以及充电动态过程。结果表明,新方法得到的现象与常规恒电势方法存在显著不同——其揭示的微分电容与界面结构与实验定量吻合,充电过程与理论吻合,从而解决了常规恒电势方法无法准确模拟金属-水溶液界面的难题。同时,该工作通过有效结合第一性原理计算和分子动力学模拟的优点,实现了对百万原子量级的双电层体系的分子模拟,其计算效率媲美常规恒电势方法。此外,该工作对于界面结构以及充电过程的分子尺度机理剖析有助于理解其他电化学领域,如电池、电催化、电镀和电容去离子领域。
该工作得到了厦门大学毛秉伟、颜佳伟教授团队,国家自然科学基金项目和华中科技大学学术前沿青年团队的资助以及武汉超算中心的支持。
论文链接:https://doi.org/10.1103/PhysRevLett.134.046201