新闻网讯 9月11日,化学与化工学院游波、夏宝玉教授的最新研究成果“Intrinsic Activity Identification of Noble Metal Single‐Sites for Electrocatalytic Chlorine Evolution”在《德国应用化学》(Angew. Chem. Int. Ed.)正式刊发。
氯(Cl₂)是最重要的工业化学品之一,其生产主要依赖于氯碱工艺,其中析氯反应(CER)作为阳极反应起着至关重要的作用。在过去的半个世纪里,基于Ti基底的混合贵金属氧化物阳极(DSA,由RuO₂和IrO₂组成),已被广泛应用于商业化的电化学氯气生产过程中。然而,这些阳极中贵金属的高含量增加了工业生产成本,并且RuO₂和IrO₂也是高效的析氧反应(OER)催化剂,它们不可避免地降低了CER的选择性。单原子催化剂(SACs)以其极高的原子利用效率在CER中展现出了巨大的应用潜力。迄今为止,科研人员已成功合成了基于Pt、Ir和Ru的各种高效的SACs电催化剂。然而,由于这些SACs在金属配位、基质宿主、金属组分以及纳米结构等结构上不同,同时电化学测量条件也缺乏统一的标准,因此客观评估这些已报道SACs的内在CER活性仍面临挑战。这些局限性严重阻碍了建立SACs结构与CER活性之间精确相关性的进程,而这种相关性对于合理设计和合成适用于氯碱工艺的新型SACs,以实现经济高效的Cl₂生成至关重要。
有鉴于此,我们成功合成了三种具有相同M–N₄配位的单原子催化剂,实现了对其本征CER活性的客观评估。电化学实验、原位Raman以及准原位EPR等研究手段表明三个M1–N–C催化剂均通过Cl介导Vomer-Heyrovský机理进行反应,并且催化反应的活性趋势为Pt1–N–C> Ir1–N–C> Ru1–N–C。通过DFT计算揭示了Cl*–Cl中间体在活性中心上的结合能是决定CER活性的关键因素。

图1.Pt1–N–C, Ir1–N–C, 和Ru1–N–C催化剂形貌表征
M1–N–C是通过将原位生长在酸化碳纳米管上贵金属掺杂的ZIF-8热解得到的。如图1a-c所示,M1–N–C前驱体在碳纳米管上呈现出具有丰富多面体晶体的链状结构。在900℃热解后,得到的M1–N–C继承了前驱体的整体管状形貌,表面的多面体晶体消失转化为包裹在碳纳米管表面的碳层。同时,球差电镜和EDX mapping (图1d-i) 证明了单原子催化剂的成功制备。
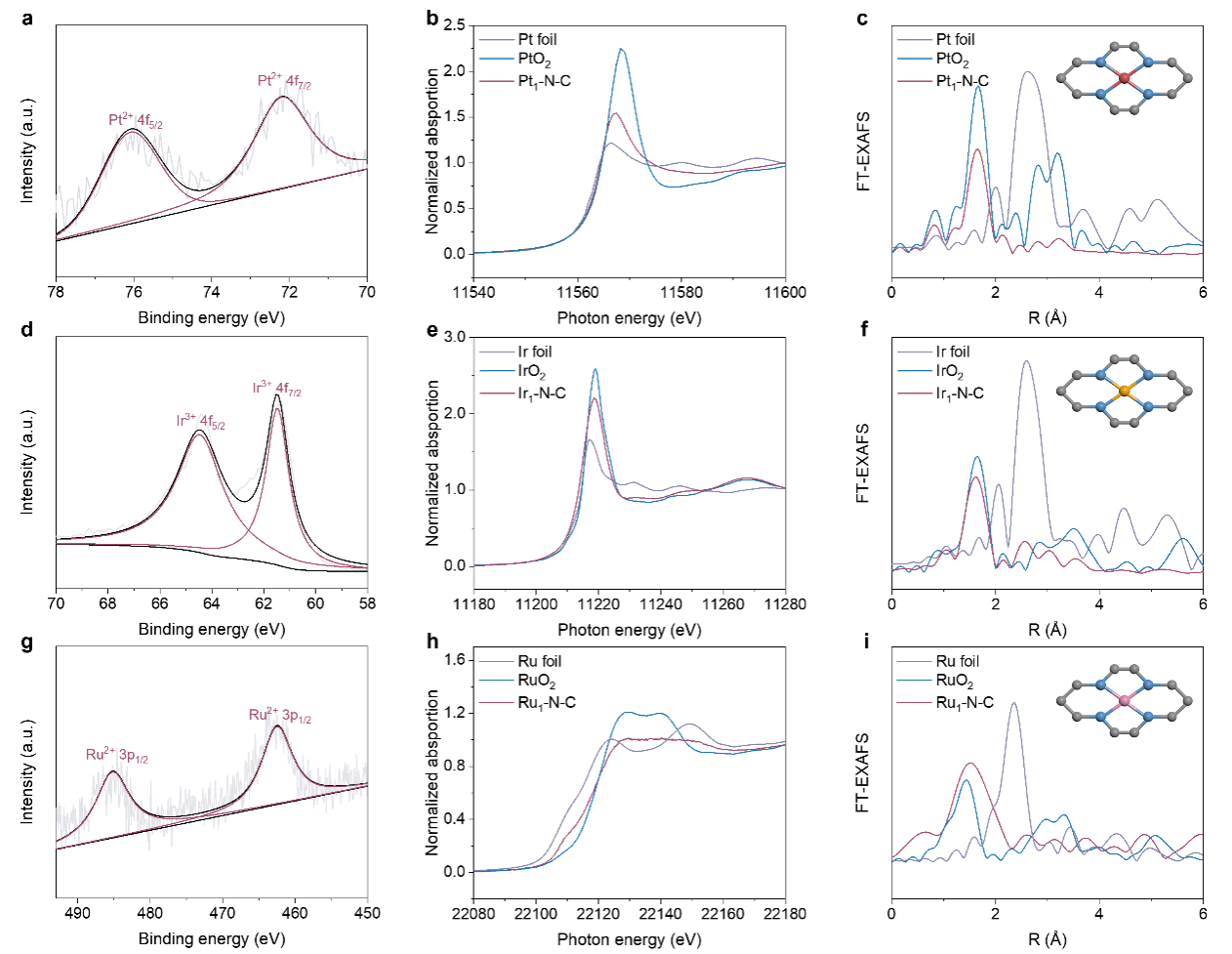
图2.Pt1–N–C, Ir1–N–C, 和Ru1–N–C催化剂电子结构表征
XPS和同步辐射X射线吸收能谱表明,M1–N–C单原子催化剂中贵金属M–N4的形式原子级分散并嵌入在碳纳米管表面的多孔氮掺杂碳中(图2)。
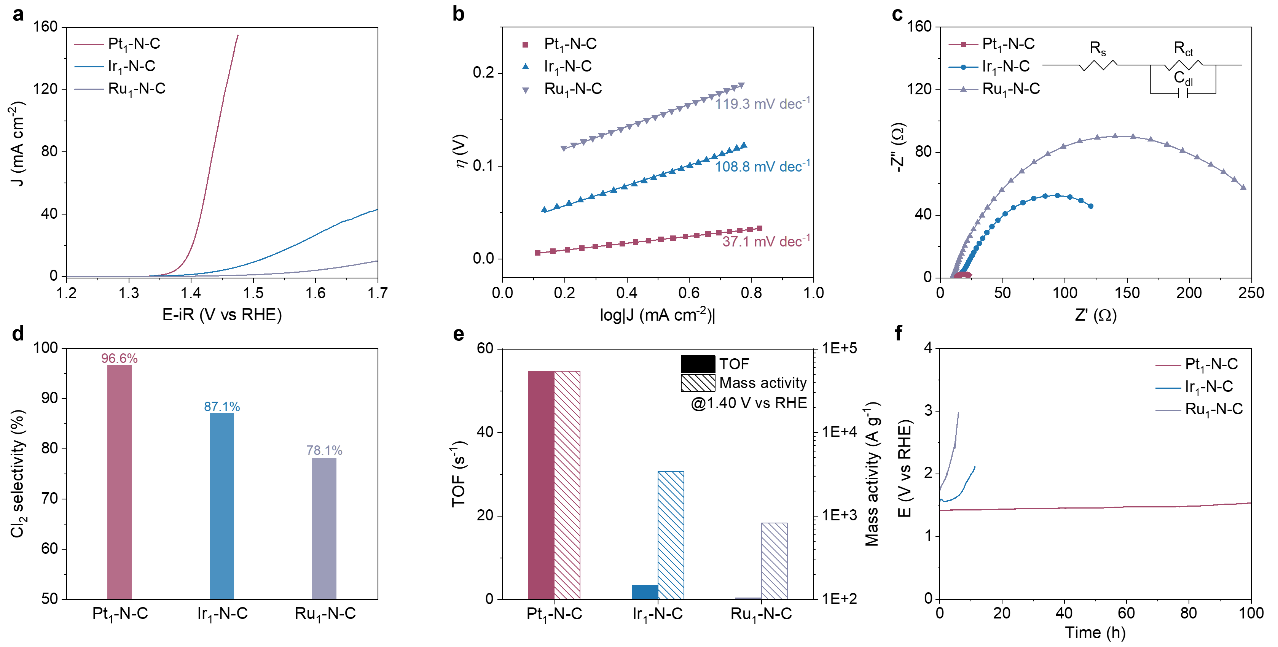
图3.Pt1–N–C, Ir1–N–C, 和Ru1–N–C的CER性能
在0.1 M HClO4和1 M NaCl的电解液中,M1–N–CSAC的CER活性呈现出Pt1–N–C> Ir1–N–C> Ru1–N–C的趋势(图3a)。例如,要达到10 mA cm-2,Pt1–N–C只需要40 mV的过电位,远远低于Ir1–N–C 和Ru1–N–C。塔菲尔斜率, 电化学阻抗谱, Cl2选择性, TOF和质量活性也证实了M1–N–CSAC 的 CER 活性顺序(图3b-e)。此外,Pt1–N–C具有优异的稳定性,在100小时内没有明显的活性衰减,而Ir1–N–C在10小时内明显失活,Ru1–N–C在5小时内明显失活(图3f)。
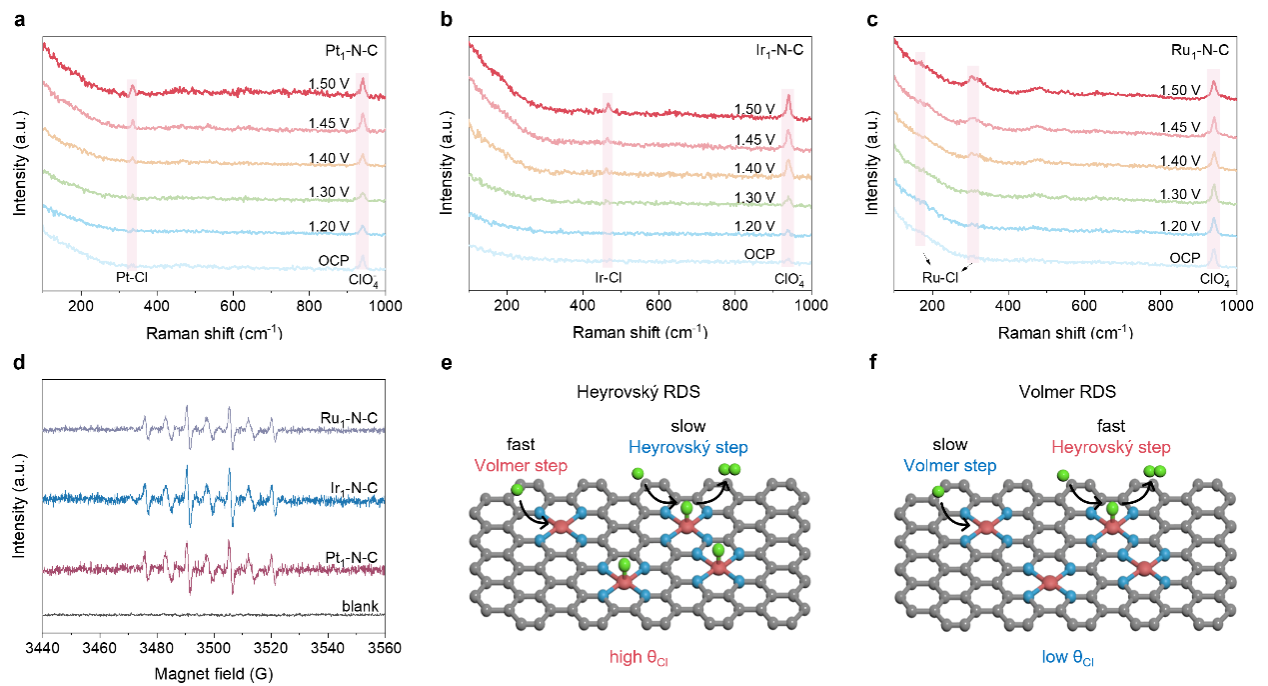
图4.M1–N–C的CER机理研究
M1–N–C上发生的反应遵循以Cl物种为中间体的Volmer-Heyrovský步骤。此外通过测量M1–N–C在不同Cl−浓度下的CER活性,推断Heyrovský为决速步(图4)。
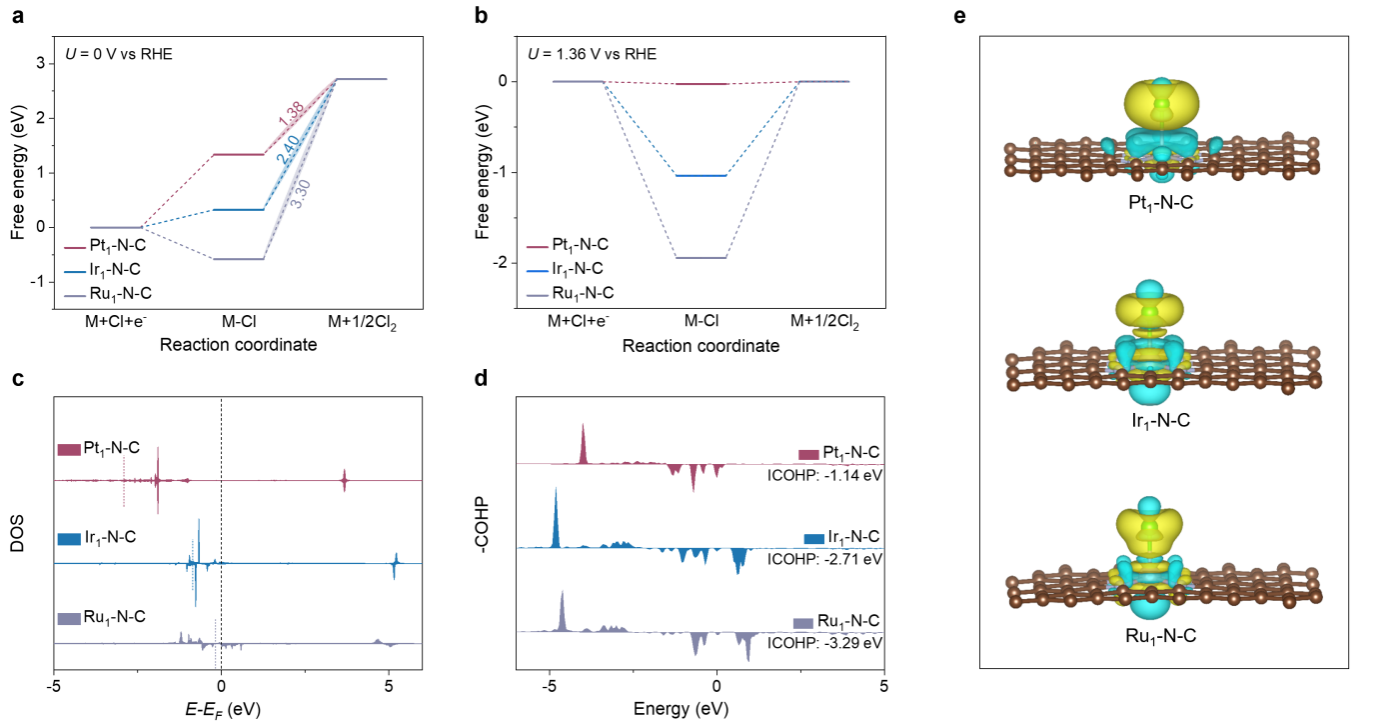
图5.DFT计算
在U=0时,Heyrovský步骤(ΔGCl*-Cl)的吉布斯自由能变化比Volmer步骤(ΔGCl*)高,这表明Heyrovský步骤是三个M-N4位点上的决速步(图5a)。此外,Pt–N4上的ΔGCl*-Cl远低于Ir-N4和Ru-N4上的ΔGCl*-Cl,这表明CER活性的顺序为Pt–N4> Ir–N4> Ru–N4。同时,Pt–N4、Ir–N4和Ru–N4在1.36 V vs RHE时的ΔGCl*-Cl分别为0.02、1.04和1.94 eV。值得注意的是,Pt–N4位点的ΔGCl*-Cl(0.02 eV)最接近于零,Pt–N4是理想催化活性位点。此外,DOS和COHP表明,Pt–N4的d带中心最负,导致其占据最高的反键态,且Pt–N4对Cl物种的吸附最弱,从而获得最小的结合能和吉布斯自由能垒。同时,Bader电荷的定量计算表明,与Ir–N4和Ru–N4相比,Pt–N4的电子转移量最大,这表明Pt–N4的反应速度最快。综上所述,这些DFT计算共同验证了Pt–N4> Ir–N4> Ru–N4的固有CER活性趋势,与实验结果相符。
综上所述,本研究成功合成了三种贵金属-氮-碳(M1–N–C)单原子催化剂,并客观评估了它们在电催化析氯反应中的本征活性。通过一系列物理化学表征和电化学实验,证实了Pt1–N–C、Ir1–N–C和Ru1–N–C具有相似的M–N4结构和CER机制,使得可以直接比较它们的活性。DFT计算表明,Pt1–N–C中活性位点Pt–N4因其最接近零的ΔGCl*–Cl值而展现出最优异的CER性能,这为高效CER电催化剂的设计提供了新的思路。
游波教授和夏宝玉教授为共同通讯作者,化学与化工学院博士研究生全力为第一作者。该研究工作得到了国家重点研发计划(2021YFA1600800)、华中科技大学学术前沿青年团队计划等项目资助。
论文链接:https://onlinelibrary.wiley.com/doi/epdf/10.1002/anie.202414202